Load necessary packages
We first load R packages required for the example here.
library(patchwork)
library(ggtree)
# spatialCCC package
library(spatialCCC)
#> Warning: replacing previous import 'S4Arrays::makeNindexFromArrayViewport' by
#> 'DelayedArray::makeNindexFromArrayViewport' when loading 'SummarizedExperiment'
#> Warning: replacing previous import 'S4Arrays::makeNindexFromArrayViewport' by
#> 'DelayedArray::makeNindexFromArrayViewport' when loading 'HDF5Array'Ligand-Receptor Database
Load built-in LRdb
This LRdb is downloaded from CellTalkDB: [http://tcm.zju.edu.cn/celltalkdb/download.php]. You can see the detail by:
?LRdb_human
?LRdb_mouseWe also created getter functions, get_LRdb() and get_LRdb_small(), to retrieve the database.
set.seed(100)
LRdb_m <-
get_LRdb("mouse", n_samples = 100)
LRdb_m %>%
dplyr::arrange(ligand_gene_symbol, receptor_gene_symbol)
#> # A tibble: 100 × 10
#> LR ligand_gene_symbol receptor_gene_symbol ligand_gene_id receptor_gene_id
#> <chr> <chr> <chr> <dbl> <dbl>
#> 1 Adam… Adam10 Notch1 11487 18128
#> 2 Adam… Adam12 Epha1 11489 13835
#> 3 Adam… Adam17 Aplp2 11491 11804
#> 4 Adam… Adam2 Itgb7 11495 16421
#> 5 Adcy… Adcyap1 Adcyap1r1 11516 11517
#> 6 Adcy… Adcyap1 Ramp3 11516 56089
#> 7 Adip… Adipoq Adipor1 11450 72674
#> 8 Adm2… Adm2 Calcrl 223780 54598
#> 9 Apoe… Apoe Ldlr 11816 16835
#> 10 Apoe… Apoe Trem2 11816 83433
#> # ℹ 90 more rows
#> # ℹ 5 more variables: ligand_ensembl_protein_id <chr>,
#> # receptor_ensembl_protein_id <chr>, ligand_ensembl_gene_id <chr>,
#> # receptor_ensembl_gene_id <chr>, evidence <chr>Load Visium spatial transcriptomic data
data_dir <- spatialCCC_example("example")
spe_brain <-
SpatialExperiment::read10xVisium(samples = data_dir,
type = "HDF5",
data = "filtered")
# to keep track of cell IDs
# spe_brain[["cell_id"]] <- colnames(spe_brain)
# Log-Normalize
spe_brain <- scater::logNormCounts(spe_brain) Cell cluster data
Now, let’s read cell cluster data, obtained using GraphST.
cell_clusters <-
read.csv(file.path(data_dir, "outs", "graphST.csv"), row.names = 1)
# Make sure the rows of cell_clusters are in the same order of spe_brain.
cell_clusters <- cell_clusters[colnames(spe_brain), ]
cluster_ids <- colnames(cell_clusters)
cluster_ids <- cluster_ids[grep("mclust_", cluster_ids)]
for (cid in cluster_ids) {
spe_brain[[cid]] <- factor(cell_clusters[[cid]])
}
plot_spatial_feature(spe = spe_brain, feature = "mclust_8")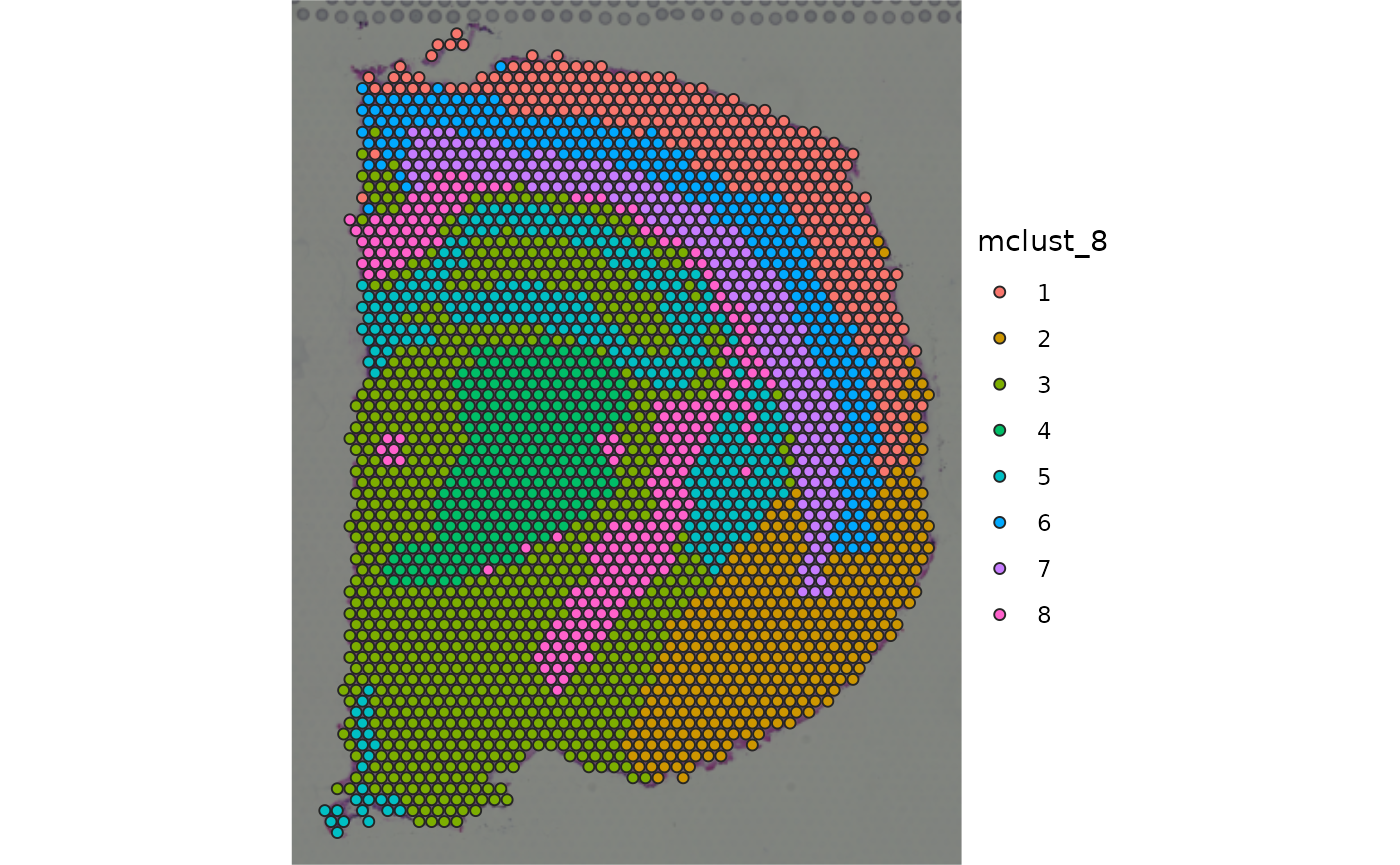
Compute Cell-Cell Communications over ligand-receptor pairs
tictoc::tic()
ccc_graph_list <-
compute_spatial_ccc_graph_list(spe = spe_brain,
assay_name = "logcounts",
LRdb = LRdb_m)
tictoc::toc()
#> 694.279 sec elapsed
ccc_graph_list <-
ccc_graph_list %>%
purrr::map(function(ccog) {
ccog %>%
add_node_annot_to_edges(c("mclust_8", "mclust_9", "mclust_10"))
})
ccc_tbl <-
ccc_graph_list %>%
purrr::map(function(ccog) {
ccog %>%
tidygraph::activate("edges") %>%
as_tibble()
}) %>%
dplyr::bind_rows(.id = "LR")Summarize cell-cell communication between cell clusters
ccc_tbl_between_clusters <-
ccc_tbl %>%
dplyr::group_by(mclust_8_src, mclust_8_dst, LR) %>%
dplyr::summarise(n = dplyr::n(),
LRscore.sum = sum(LRscore),
.groups = "drop")
ccc_tbl_between_clusters %>%
dplyr::arrange(desc(n))
#> # A tibble: 1,327 × 5
#> mclust_8_src mclust_8_dst LR n LRscore.sum
#> <fct> <fct> <chr> <int> <dbl>
#> 1 3 3 App_Lrp1 4480 3576.
#> 2 3 3 Nlgn2_Nrxn1 3592 2585.
#> 3 3 3 Ncam1_Fgfr1 3093 2167.
#> 4 3 3 S100a1_Ryr2 2349 1592.
#> 5 2 2 App_Lrp1 2136 1710.
#> 6 3 3 Apoe_Ldlr 1953 1487.
#> 7 2 2 Nlgn2_Nrxn1 1840 1386.
#> 8 1 1 App_Lrp1 1742 1406.
#> 9 3 3 Apoe_Trem2 1720 1322.
#> 10 3 3 Adcyap1_Adcyap1r1 1687 1131.
#> # ℹ 1,317 more rows
ccc_tbl_between_clusters %>%
dplyr::filter(mclust_8_dst != mclust_8_src) %>%
dplyr::arrange(desc(n))
#> # A tibble: 1,004 × 5
#> mclust_8_src mclust_8_dst LR n LRscore.sum
#> <fct> <fct> <chr> <int> <dbl>
#> 1 3 5 App_Lrp1 304 241.
#> 2 5 3 App_Lrp1 300 238.
#> 3 5 3 Nlgn2_Nrxn1 255 184.
#> 4 3 5 S100a1_Ryr2 224 161.
#> 5 8 3 App_Lrp1 212 171.
#> 6 3 5 Ncam1_Fgfr1 208 145.
#> 7 3 8 App_Lrp1 199 157.
#> 8 3 5 Nlgn2_Nrxn1 195 141.
#> 9 5 3 Ncam1_Fgfr1 165 116.
#> 10 3 5 Calm2_Cacna1c 163 122.
#> # ℹ 994 more rowsConvert CCC table to CCC graph
summarize_ccc_graph_metrics() summarize those graph metrics for each LR pair.
tictoc::tic()
ccc_graph_metrics_summary_df <-
summarize_ccc_graph_metrics(ccc_graph_list)
tictoc::toc()
#> 0.237 sec elapsed
ccc_graph_metrics_summary_df %>%
dplyr::arrange(graph_component_count)
#> # A tibble: 41 × 12
#> LR graph_n_nodes graph_n_edges graph_component_count graph_motif_count
#> <chr> <int> <dbl> <dbl> <int>
#> 1 App_Lrp1 2700 17430 3 27811
#> 2 Ncam1_Fg… 2549 9640 3 18340
#> 3 Nlgn2_Nr… 2675 14289 3 25062
#> 4 S100a1_R… 2591 10689 5 20094
#> 5 Calm2_Ca… 2461 8105 6 16288
#> 6 Apoe_Tre… 2560 6937 8 14484
#> 7 Apoe_Ldlr 2295 5608 26 11724
#> 8 Slit1_Ro… 1651 4373 41 7866
#> 9 Calm2_My… 1967 4247 42 8830
#> 10 Adcyap1_… 1642 4172 43 7801
#> # ℹ 31 more rows
#> # ℹ 7 more variables: graph_diameter <dbl>, graph_un_diameter <dbl>,
#> # graph_mean_dist <dbl>, graph_circuit_rank <dbl>, graph_reciprocity <dbl>,
#> # graph_clique_num <int>, graph_clique_count <int>summarize_ccc_graph_metrics(…, level = “group”) summarizes the metrics for each subgraph (group) in CCC graph (LR)
tictoc::tic()
ccc_graph_group_metrics_summary_df <-
summarize_ccc_graph_metrics(ccc_graph_list, level = "group")
tictoc::toc()
#> 0.305 sec elapsed
ccc_graph_group_metrics_summary_df
#> # A tibble: 2,877 × 12
#> LR group group_n_nodes group_n_edges group_adhesion group_motif_count
#> <chr> <int> <int> <dbl> <dbl> <int>
#> 1 Rspo3_Lgr6 38 2 1 0 0
#> 2 Rspo3_Lgr6 13 3 3 0 1
#> 3 Rspo3_Lgr6 39 2 1 0 0
#> 4 Rspo3_Lgr6 14 3 2 0 1
#> 5 Rspo3_Lgr6 5 4 3 0 3
#> 6 Rspo3_Lgr6 6 4 4 0 3
#> 7 Rspo3_Lgr6 75 1 1 0 0
#> 8 Rspo3_Lgr6 40 2 1 0 0
#> 9 Rspo3_Lgr6 41 2 1 0 0
#> 10 Rspo3_Lgr6 15 3 3 0 1
#> # ℹ 2,867 more rows
#> # ℹ 6 more variables: group_diameter <dbl>, group_un_diameter <dbl>,
#> # group_mean_dist <dbl>, group_girth <dbl>, group_circuit_rank <dbl>,
#> # group_reciprocity <dbl>Visualization
LR_of_interest <- "Pdgfb_Pdgfra"
ccc_graph_list[[LR_of_interest]] %>%
tidygraph::activate(edges) %>%
tibble::as_tibble()
#> # A tibble: 2,406 × 40
#> from to src dst d norm.d ligand receptor LRscore weight WLRscore
#> <int> <int> <chr> <chr> <dbl> <dbl> <chr> <chr> <dbl> <dbl> <dbl>
#> 1 1 1 AAACA… AAAC… 0 0 Pdgfb Pdgfra 0.621 1 0.621
#> 2 1 920 AAACA… AGAG… 138. 1.01 Pdgfb Pdgfra 0.649 0.980 0.636
#> 3 1 921 AAACA… GGCA… 138. 1.01 Pdgfb Pdgfra 0.684 0.987 0.675
#> 4 1 896 AAACA… TTCT… 138. 1.01 Pdgfb Pdgfra 0.629 0.980 0.616
#> 5 2 922 AAACA… TCAC… 138. 1.01 Pdgfb Pdgfra 0.599 0.980 0.587
#> 6 3 3 AAACC… AAAC… 0 0 Pdgfb Pdgfra 0.541 1 0.541
#> 7 3 923 AAACC… CGCT… 138 1.01 Pdgfb Pdgfra 0.631 0.986 0.622
#> 8 3 924 AAACC… TTGG… 138. 1.01 Pdgfb Pdgfra 0.509 0.980 0.498
#> 9 4 4 AAACG… AAAC… 0 0 Pdgfb Pdgfra 0.641 1 0.641
#> 10 4 50 AAACG… AAGA… 138 1.01 Pdgfb Pdgfra 0.670 0.986 0.660
#> # ℹ 2,396 more rows
#> # ℹ 29 more variables: LRscore_perc_rank <dbl>, graph_n_nodes <int>,
#> # graph_n_edges <dbl>, graph_component_count <dbl>, graph_motif_count <int>,
#> # graph_diameter <dbl>, graph_un_diameter <dbl>, graph_mean_dist <dbl>,
#> # graph_circuit_rank <dbl>, graph_reciprocity <dbl>, graph_clique_num <int>,
#> # graph_clique_count <int>, group <int>, group_n_nodes <int>,
#> # group_n_edges <dbl>, group_adhesion <dbl>, group_motif_count <int>, …spatial CCC graph plot with tissue image
gp_spccc <-
plot_spatial_ccc_graph(
ccc_graph = ccc_graph_list[[LR_of_interest]],
tissue_img = SpatialExperiment::imgRaster(spe_brain),
node_color = "mclust_8",
node_size = 1,
node_alpha = 0.5,
edge_color = "group",
# clip = TRUE,
which_on_top = "edge"
)
gp_spccc_0 <-
plot_spatial_feature(spe = spe_brain,
feature = "mclust_8")
wrap_plots(gp_spccc, gp_spccc_0, ncol = 2)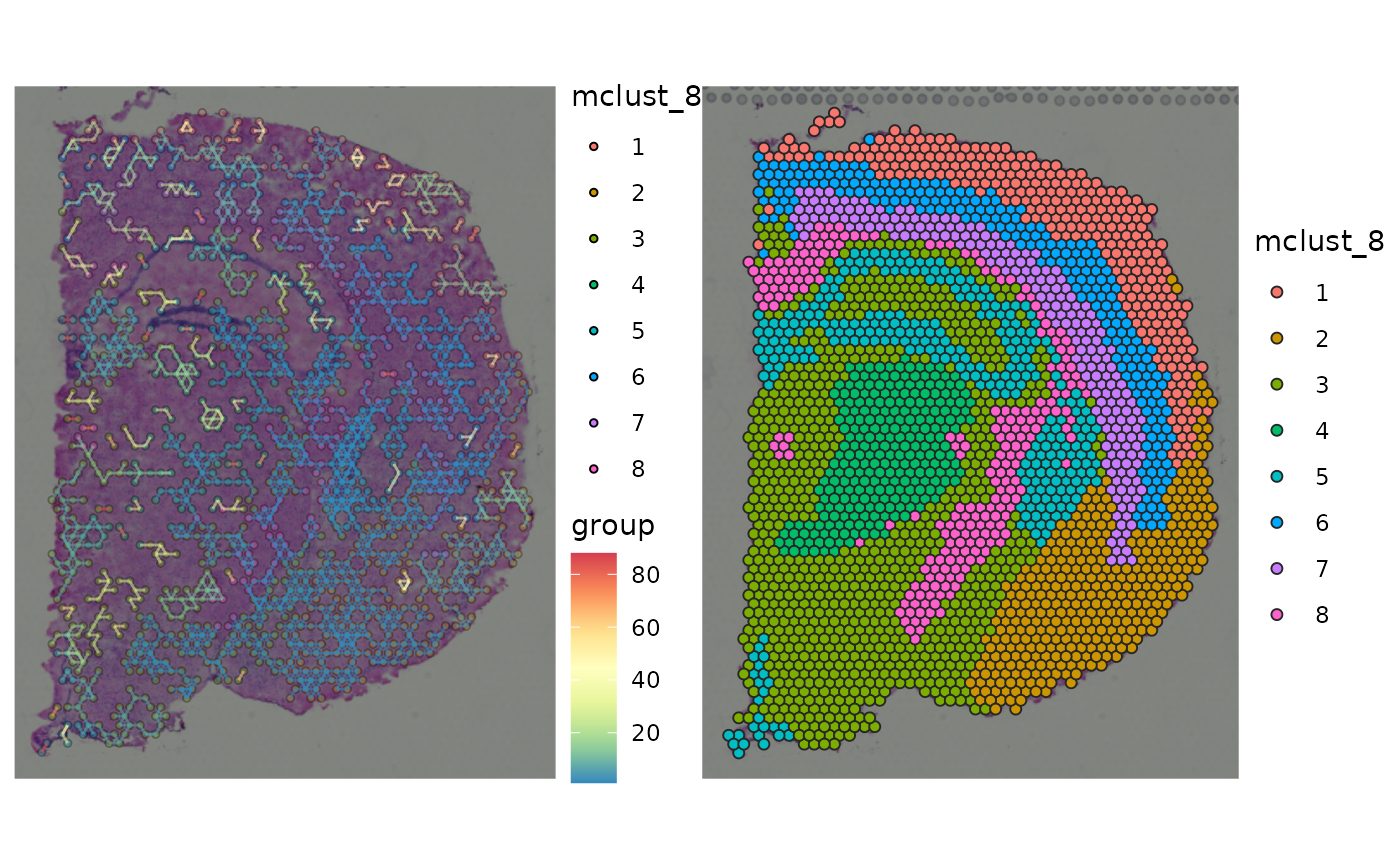
ccc_graph_temp <-
ccc_graph_list[[LR_of_interest]] %>%
tidygraph::activate("edges") %>%
tidygraph::filter(mclust_8_src != mclust_8_dst) %>%
tidy_up_ccc_graph()
cells_of_interest <-
ccc_graph_temp %>%
tidygraph::activate("nodes") %>%
dplyr::pull("name")
gp_spccc <-
plot_spatial_ccc_graph(
ccc_graph = ccc_graph_temp,
tissue_img = SpatialExperiment::imgRaster(spe_brain),
node_color = "n.inflow",
node_size = 1.25,
node_alpha = 1,
edge_color = "group",
show_arrow = TRUE,
# clip = TRUE,
which_on_top = "node"
)
gp_spccc_0 <-
plot_spatial_ccc_graph(
ccc_graph = ccc_graph_list[[LR_of_interest]],
tissue_img = SpatialExperiment::imgRaster(spe_brain),
image_alpha = 0,
cells_of_interest = cells_of_interest,
edges_expanded_to_group = FALSE,
node_color = "mclust_8",
node_size = 1.25,
node_alpha = 0.5,
edge_color = "group",
show_arrow = TRUE,
# clip = TRUE,
which_on_top = "node"
)
gp_spccc_1 <-
plot_spatial_feature(spe = spe_brain,
feature = "mclust_8",
cells_of_interest = cells_of_interest)
wrap_plots(gp_spccc, gp_spccc_1, ncol = 2)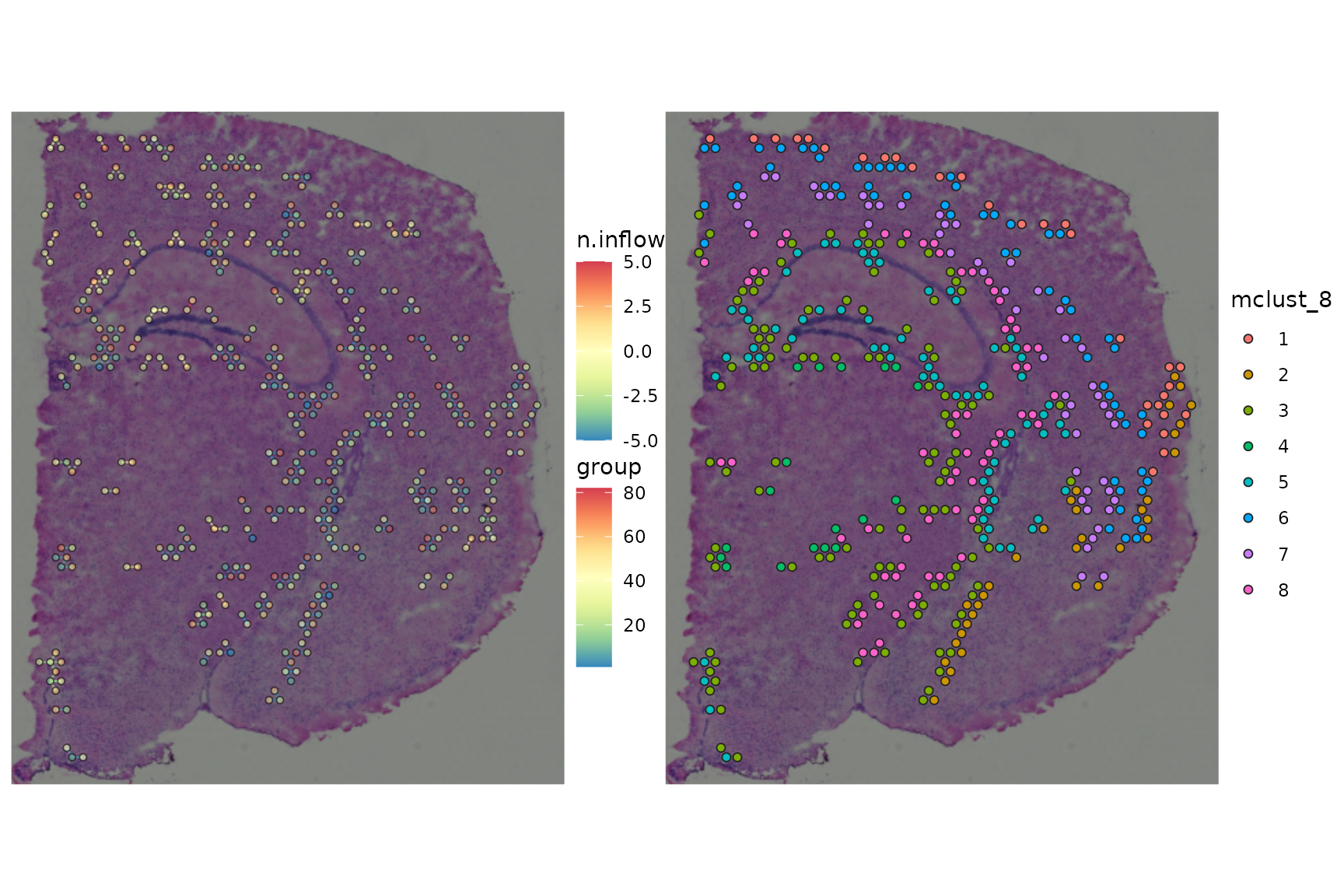
gp_spccc_0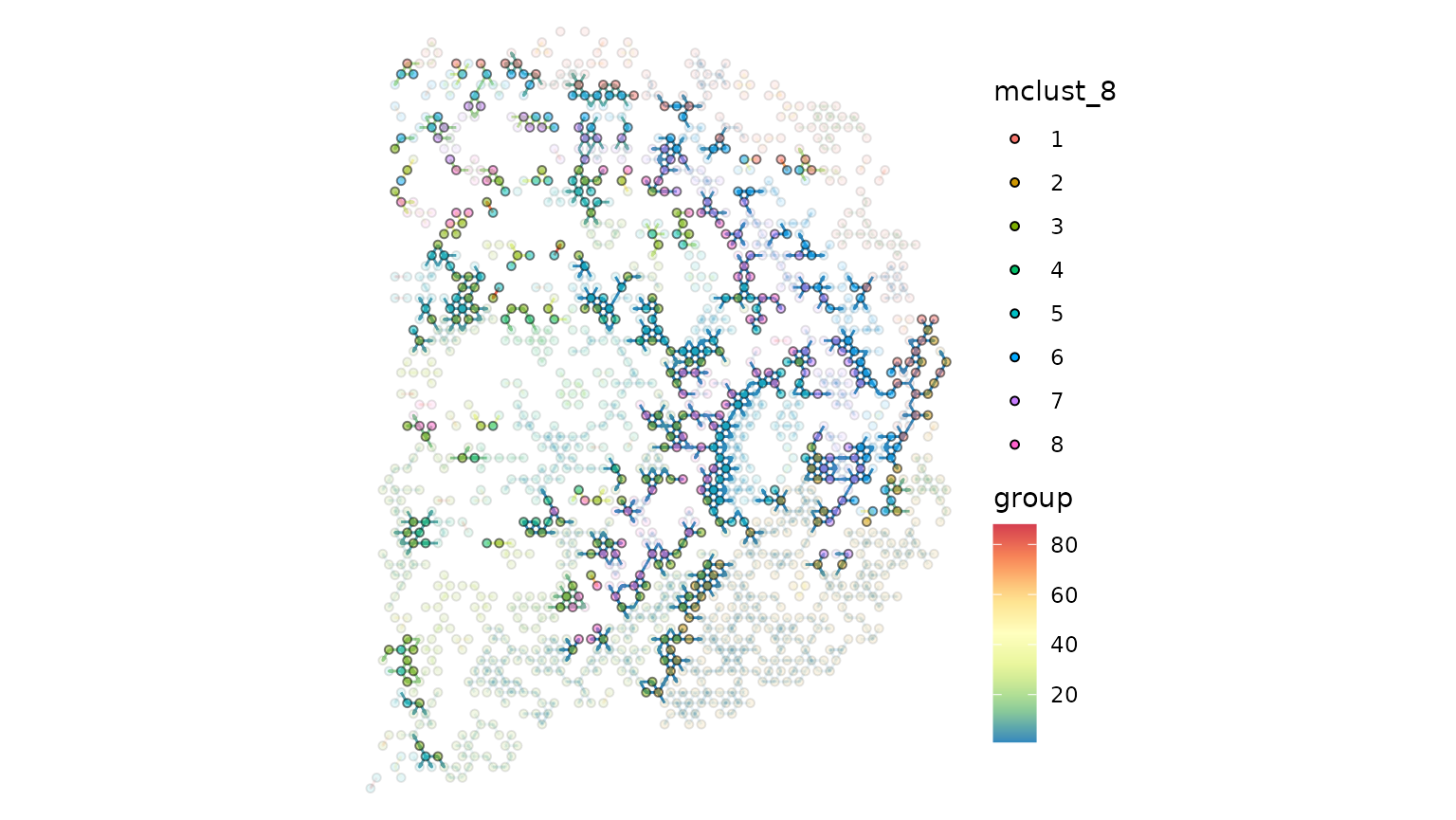
spatial CCC graph plot without tissue image
In this case, graph layout can be “spatial” which keeps the original spatial locations, or other graph layout algorithm supported by igraph package.
gp_spccc <-
plot_spatial_ccc_graph(
ccc_graph = ccc_graph_list[[LR_of_interest]],
graph_layout = "spatial",
node_color = "mclust_8",
node_size = 1,
edge_color = "group_diameter",
clip = TRUE,
# ghost_img = TRUE,
which_on_top = "edge"
)
gp_spccc_0 <-
plot_spatial_ccc_graph(
ccc_graph =
ccc_graph_list[[LR_of_interest]],
graph_layout = "spatial",
node_color = "mclust_8",
node_size = 1,
edge_color = "group_diameter",
clip = TRUE,
# ghost_img = TRUE,
which_on_top = "node"
)
wrap_plots(gp_spccc, gp_spccc_0, ncol = 2, guides = "collect")
Below uses “auto” layout (“kk” spring layout).
plot_spatial_ccc_graph(ccc_graph = ccc_graph_list[[LR_of_interest]],
# tissue_img = SpatialExperiment::imgRaster(spe_brain),
node_color = "group",
node_size = 0.1,
edge_color = "group_diameter",
edge_width = 0.1,
which_on_top = "edge")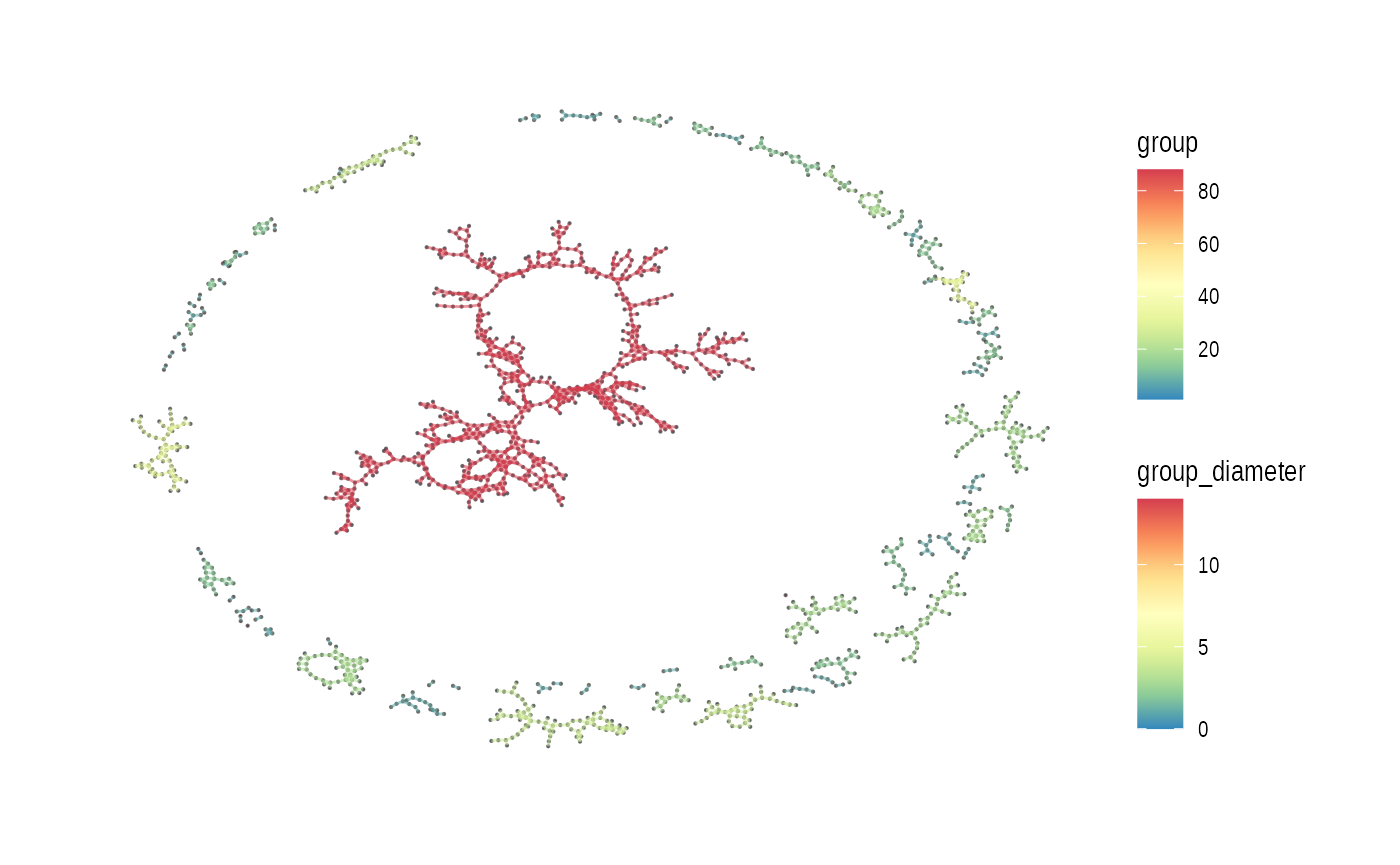
In this case, below is “stress” layout.
plot_spatial_ccc_graph(ccc_graph = ccc_graph_list[[LR_of_interest]],
# tissue_img = SpatialExperiment::imgRaster(spe_brain),
graph_layout = "stress",
node_color = "group",
edge_color = "group_diameter",
edge_width = 0.25,
which_on_top = "edge")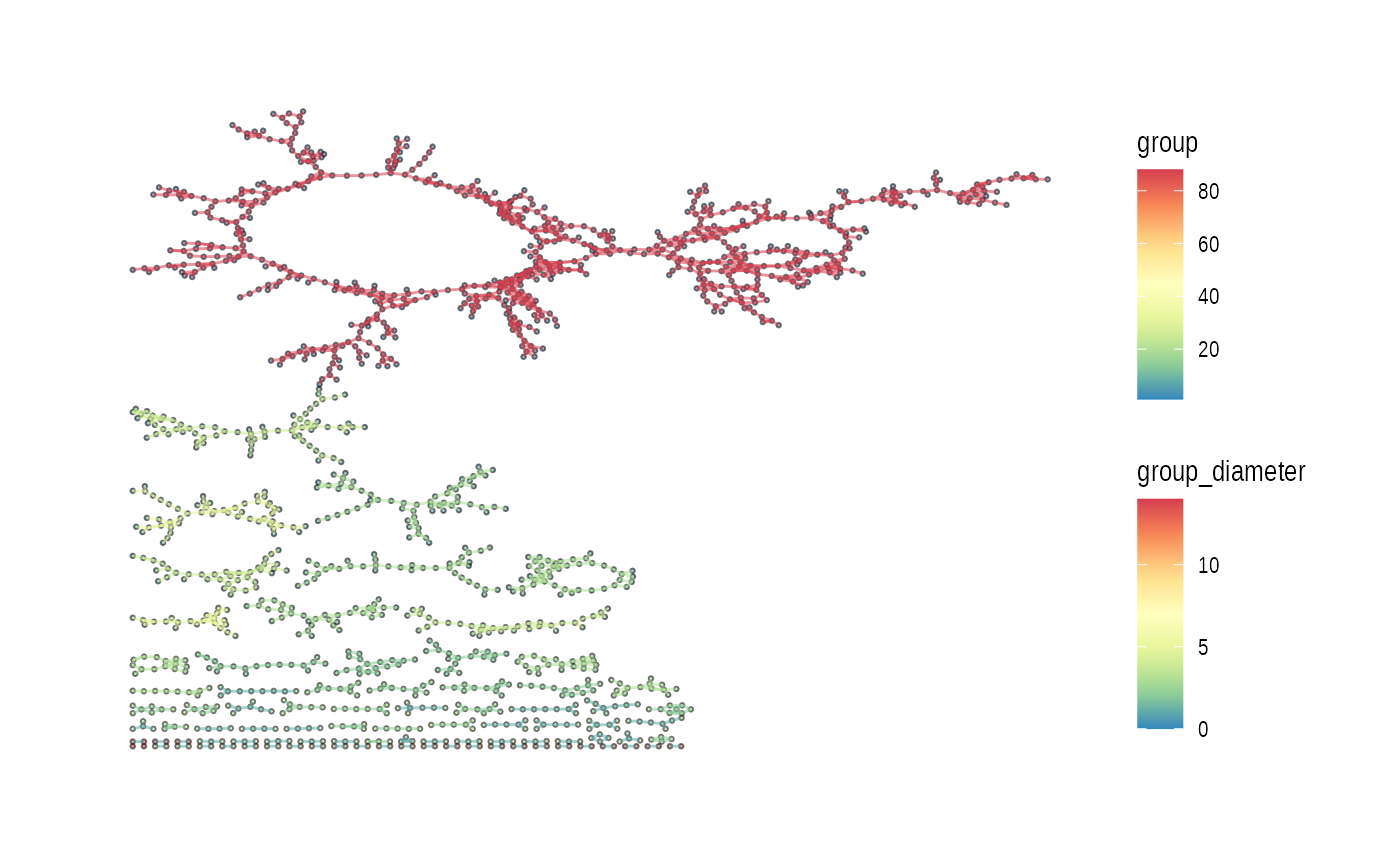
Cell-overlap distance
tictoc::tic()
cell_overlap_dist <-
dist_cell_overlap_ccc_tbl(ccc_tbl)
tictoc::toc()
#> 0.878 sec elapsed
tictoc::tic()
cell_overlap_lf <-
lf_cell_overlap_ccc_tbl(ccc_tbl)
tictoc::toc()
#> 1.229 sec elapsed
tictoc::tic()
LRs_high_cell_overlap <-
cell_overlap_lf %>%
dplyr::filter(d < 1) %>%
dplyr::select(lr1, lr2) %>%
unlist() %>% unique()
tictoc::toc()
#> 0.011 sec elapsed
tictoc::tic()
high_cell_overlap_dist <-
cell_overlap_dist[LRs_high_cell_overlap, LRs_high_cell_overlap]
tictoc::toc()
#> 0.003 sec elapsed
tictoc::tic()
high_cell_overlap_dist2 <-
cell_overlap_lf %>%
dplyr::filter(d < 1) %>%
dplyr::select(lr1, lr2, d) %>%
lf_to_dist()
tictoc::toc()
#> 0.031 sec elapsed
LR_ccc_summary_tbl <-
ccc_tbl %>%
dplyr::pull(LR) %>%
table() %>%
tibble::as_tibble() %>%
dplyr::rename("LR" = ".") %>%
dplyr::arrange(desc(n)) %>%
dplyr::left_join(
ccc_tbl %>%
dplyr::select(LR, ligand, receptor) %>%
dplyr::distinct(),
by = "LR"
)
ape::as.phylo(hclus.res) %>%
ggtree(layout="dendrogram") %<+% LR_ccc_summary_tbl +
aes(color=receptor) +
theme(legend.position = "none") +
geom_tippoint(aes(size=n), alpha=0.5) +
geom_tiplab(size=2, offset=-0.15) +
xlim_tree(3)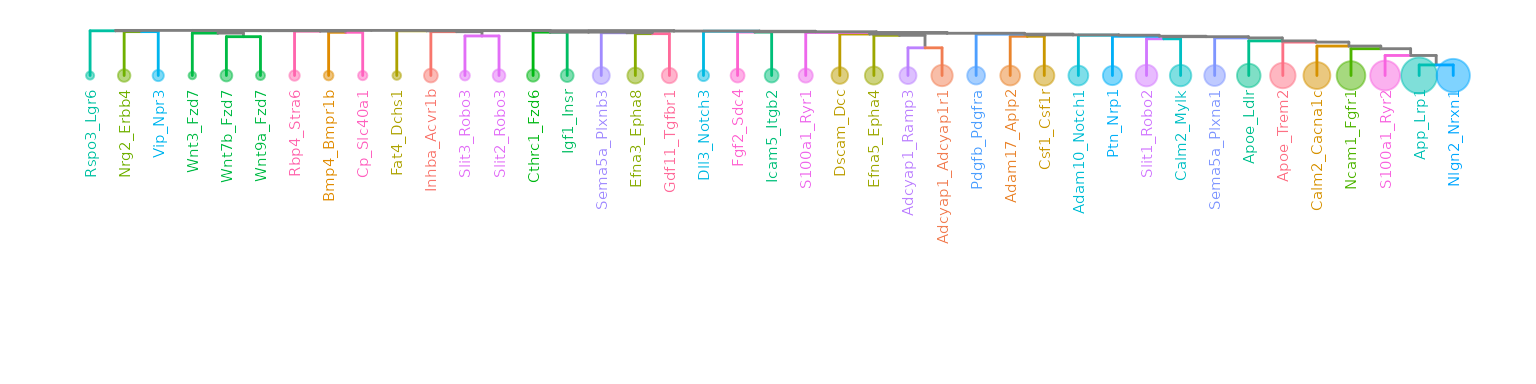
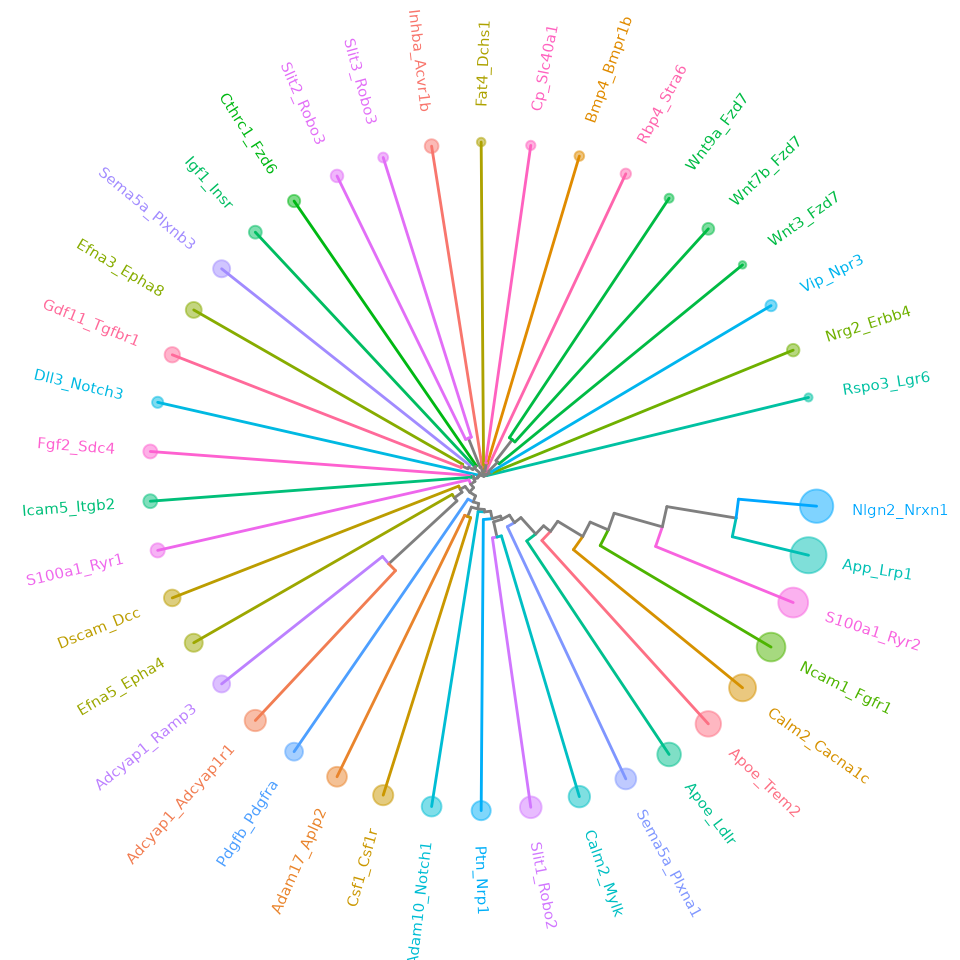
ape::as.phylo(hclus.res) %>%
ggtree(layout = "circular") %<+% LR_ccc_summary_tbl +
aes(color=receptor) +
# aes(color=ligand) +
theme(legend.position = "none") +
geom_tippoint(aes(size=n), alpha=0.5) +
geom_tiplab(size=2, offset=0.05)